
My name is Hannes Röst and I am a bioinformatics researcher interested in complex molecular phenomena which I investigate using multiple computational and statistical methods. I am studying how static and dynamic molecular traits relate to observable and disease related phenotypes. In addition, I have broad interests in utilitarianism, education, learning and rationality. I am a native of Switzerland and studied Biochemistry and Bioinformatics at ETH Zurich (Switzerland) and UC Berkeley (exchange semester). I have completed my PhD in computational biology and mass spectrometry at ETH Zurich (Switzerland) under the supervision of Dr. Lars Malmström and Prof. Ruedi Aebersold in December 2014 and a post-doctoral fellowship working with Prof. Mike Snyder at Stanford University in 2017. I am currently an Assistant Professor at the University of Toronto.
Since the sequencing of the first complete human genome, a wealth of information has been available to biologists, transforming (molecular) biology to become more quantitative, less biased and allowing for data-driven research. Using high-throughput experimental methods allows researchers to sequence complete genomes and monitor the expression of hundreds to thousands of genes in a single experiment. To process this wealth of data, we have to develop novel computational and statistical methods which do not yet exist. The challenge is to use appropriate signal processing techniques to separate signal from noise, accurately determine the quality of the data and then combine heterogeneous datasets in order to better understand complex biological systems.
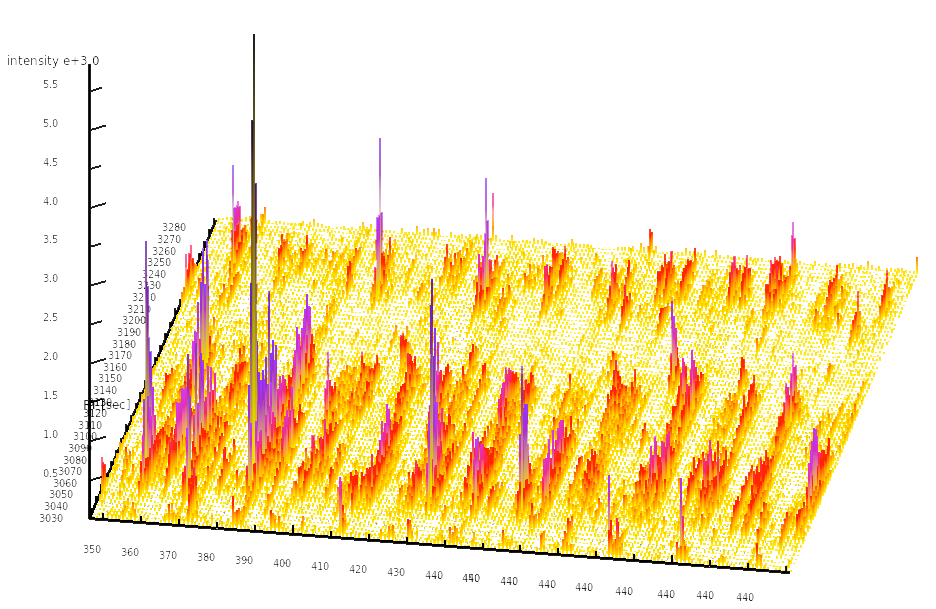
Among the quantitative high-throughput methods currently available to biology researchers, mass spectrometry-based proteomics stands out in its ability to accurately measure the quantities of hundreds to thousands of proteins in a single sample. In the Aebersold lab, we have developed a targeted proteomics strategy termed SWATH-MS which is based on data-independent acquisition and is used in conjunction with novel data analysis algorithms to extract, identify and quantify signals in a targeted fashion from the generated data. This technology allows us to quantify analyte signals consistently and reproducibly across dozens or hundreds of samples, allowing mass spectrometry to become a viable choice for systems biology and clinical experiments.
In my research, I aim to develop and apply computational approaches to study biological systems, specifically the relationship between molecular traits on (epi-)genetic, transcriptomic and proteomic level and macroscopic phenotypes. I am interested to develop methods to more accurately infer and quantify these molecular traits and relate them observable phenotypes. To improve quantitative measurements of proteins across many biological samples, I was involved with the development of computational analysis algorithms for SWATH-MS. For more information, see the research section.
In addition to my research, I also work on a few other projects in my free time. I am a board member of Aiducation International which has sponsored the high-school education of over 900 ambitious, motivated and extremely talented young students in developing countries. I also help to edit Wikipedia and write external software for the Wikipedia community. Together with my wife Martina, we maintain a website of free and libre vegetarian recipes (chuchitisch.ch). For more information, see the personal interests section.